YOUR VISION. OUR MISSION.

RESOURCE






















OUR PRODUCTS
YOUR VISION. OUR MISSION.
OUR PRODUCTS
INDICATION(S) AND IMPORTANT SAFETY INFORMATION
Click the links to go to each product's Indication(s) and Important Safety Information.
ACUVAIL® (ketorolac tromethamine ophthalmic solution) 0.45% Important Information
INDICATION
ACUVAIL® (ketorolac tromethamine ophthalmic solution) 0.45% is a nonsteroidal anti-inflammatory indicated for the treatment of pain and inflammation following cataract surgery.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
ACUVAIL solution is contraindicated in patients with previously demonstrated hypersensitivity to any of the ingredients in the formulation.
WARNINGS AND PRECAUTIONS
Delayed Healing: Topical nonsteroidal anti-inflammatory drugs (NSAIDs) may slow or delay healing. Topical corticosteroids are also known to slow or delay healing. Concomitant use of topical NSAIDs and topical steroids may increase the potential for healing problems.
Potential for Cross-Sensitivity: There is the potential for cross-sensitivity to acetylsalicylic acid, phenylacetic acid derivatives, and other NSAIDs. Caution should be used when treating patients who have previously exhibited sensitivities to these drugs.
Increased Bleeding Time: With some NSAIDs, there exists the potential for increased bleeding time due to interference with thrombocyte aggregation. There have been reports that ocularly applied nonsteroidal anti-inflammatory drugs may cause increased bleeding of ocular tissues (including hyphemas) in conjunction with ocular surgery. ACUVAIL solution should be used with caution in patients with known bleeding tendencies or who are receiving other medications, which may prolong bleeding time.
Corneal Effects: Use of topical NSAIDs may result in keratitis. In some susceptible patients, continued use of topical NSAIDs may result in epithelial breakdown, corneal thinning, corneal erosion, corneal ulceration, or corneal perforation. These events may be sight threatening. Patients with evidence of corneal epithelial breakdown should immediately discontinue use of topical NSAIDs and should be closely monitored for corneal health.
Postmarketing experience with topical NSAIDs suggests that patients with complicated ocular surgeries, corneal denervation, corneal epithelial defects, diabetes mellitus, ocular surface diseases (eg, dry eye syndrome), rheumatoid arthritis, or repeat ocular surgeries within a short period of time may be at increased risk for corneal adverse events, which may become sight threatening.
Postmarketing experience with topical NSAIDs also suggests that use more than 1 day prior to surgery or use beyond 14 days postsurgery may increase patient risk for the occurrence and severity of corneal adverse events.
Contact Lens Wear: ACUVAIL should not be administered while wearing contact lenses.
ADVERSE REACTIONS
The most common adverse events were reported in 1% to 6% of patients and included increased intraocular pressure, conjunctival hyperemia and/or hemorrhage, corneal edema, ocular pain, headache, tearing, and vision blurred. Some of these events may be the consequence of the cataract surgical procedure.
Please see accompanying full Prescribing Information or visit https://www.rxabbvie.com/pdf/acuvail_pi.pdf
ALPHAGAN® P (brimonidine tartrate ophthalmic solution) 0.1% or 0.15% Important Information
INDICATIONS AND USAGE
ALPHAGAN® P (brimonidine tartrate ophthalmic solution) 0.1% or 0.15% is an alpha-adrenergic receptor agonist indicated for the reduction of elevated intraocular pressure (IOP) in patients with open-angle glaucoma or ocular hypertension.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
Neonates and Infants (under the age of 2 years): ALPHAGAN® P is contraindicated in neonates and infants (under the age of 2 years).
Hypersensitivity Reactions: ALPHAGAN® P is contraindicated in patients who have exhibited a hypersensitivity reaction to any component of this medication in the past.
WARNINGS AND PRECAUTIONS
Potentiation of Vascular Insufficiency: ALPHAGAN® P may potentiate syndromes associated with vascular insufficiency. ALPHAGAN® P should be used with caution in patients with depression, cerebral or coronary insufficiency, Raynaud's phenomenon, orthostatic hypotension, or thromboangiitis obliterans.
Severe Cardiovascular Disease: Although brimonidine tartrate ophthalmic solution had minimal effect on the blood pressure of patients in clinical studies, caution should be exercised in treating patients with severe cardiovascular disease.
Contamination of Topical Ophthalmic Products After Use: There have been reports of bacterial keratitis associated with the use of multiple-dose containers of topical ophthalmic products. These containers had been inadvertently contaminated by patients who, in most cases, had a concurrent corneal disease or a disruption of the ocular epithelial surface.
DRUG INTERACTIONS
Antihypertensives/Cardiac Glycosides: ALPHAGAN® P may reduce blood pressure. Use caution in patients on antihypertensives and/or cardiac glycosides.
CNS Depressants: Although specific drug interaction studies have not been conducted with ALPHAGAN® P, the possibility of an additive or potentiating effect with CNS depressants (alcohol, barbiturates, opiates, sedatives, or anesthetics) should be considered.
Tricyclic Antidepressants: Tricyclic antidepressants have been reported to blunt the hypotensive effect of systemic clonidine. It is not known whether the concurrent use of these agents with ALPHAGAN® P in humans can lead to resulting interference with the IOP-lowering effect. Use caution in patients on tricyclic antidepressants, which can affect the metabolism and uptake of circulating amines.
Monoamine Oxidase Inhibitors: Monoamine oxidase (MAO) inhibitors may theoretically interfere with the metabolism of brimonidine and potentially result in an increased systemic side effect such as hypotension. Use caution in patients on MAO inhibitors, which can affect the metabolism and uptake of circulating amines.
ADVERSE REACTIONS
Adverse reactions occurring in approximately 10% to 20% of the subjects receiving brimonidine ophthalmic solution (0.1% to 0.2%) included: allergic conjunctivitis, conjunctival hyperemia, and eye pruritus. Adverse reactions occurring in approximately 5% to 9% included: burning sensation, conjunctival folliculosis, hypertension, ocular allergic reaction, oral dryness, and visual disturbance.
Please see accompanying full Prescribing Information or visit https://www.rxabbvie.com/pdf/alphagan_pi.pdf
COMBIGAN® (brimonidine tartrate/timolol maleate ophthalmic solution) 0.2%/0.5% Important Information
INDICATIONS AND USAGE
COMBIGAN® (brimonidine tartrate/timolol maleate ophthalmic solution) 0.2%/0.5% is an alpha-adrenergic receptor agonist with a beta-adrenergic receptor inhibitor indicated for the reduction of elevated intraocular pressure (IOP) in patients with glaucoma or ocular hypertension who require adjunctive or replacement therapy due to inadequately controlled IOP; the IOP-lowering of COMBIGAN® dosed twice a day was slightly less than that seen with the concomitant administration of 0.5% timolol maleate ophthalmic solution dosed twice a day and 0.2% brimonidine tartrate ophthalmic solution dosed three times per day.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
COMBIGAN® is contraindicated in patients with reactive airway disease including bronchial asthma; a history of bronchial asthma; severe chronic obstructive pulmonary disease; in patients with sinus bradycardia; second or third degree atrioventricular block; overt cardiac failure; cardiogenic shock; in neonates and infants (aged 2 years and younger); in patients with a hypersensitivity reaction to any component of COMBIGAN® in the past.
WARNINGS AND PRECAUTIONS
COMBIGAN® contains timolol maleate. COMBIGAN® is administered topically, but can be absorbed systemically. The adverse reactions with systemic administration of beta-adrenergic blocking agents may occur with topical use (eg, severe respiratory reactions and cardiac reactions, including death due to bronchospasm in patients with asthma, and rarely death in association with cardiac failure, have been reported with systemic or ophthalmic administration of timolol maleate). Ophthalmic beta-blockers may impair compensatory tachycardia and increase risk of hypotension.
Sympathetic stimulation may be essential to support the circulation in patients with diminished myocardial contractility, and its inhibition by beta-adrenergic receptor blockade may precipitate more severe failure. In patients with no history of cardiac failure, continued depression of the myocardium with beta-blocking agents over time can lead to cardiac failure. Discontinue COMBIGAN® at the first sign or symptom of cardiac failure.
Patients with chronic obstructive pulmonary disease (eg, chronic bronchitis, emphysema) of mild or moderate severity, bronchospastic disease, or a history of bronchospastic disease should not receive beta-blocking agents, including COMBIGAN®.
COMBIGAN® may potentiate syndromes associated with vascular insufficiency. Use caution in patients with depression, cerebral or coronary insufficiency, Raynaud's phenomenon, orthostatic hypotension, or thromboangiitis obliterans.
Patients taking beta-blockers with a history of atopy or severe anaphylactic reactions to a variety of allergens may be more reactive to repeated accidental, diagnostic, or therapeutic challenge with such allergens. Such patients may be unresponsive to the usual doses of epinephrine used to treat anaphylactic reactions.
Beta-adrenergic blockade can potentiate muscle weakness with myasthenic symptoms (eg, diplopia, ptosis, and generalized weakness). Although rare, timolol can increase muscle weakness in some patients with myasthenia gravis or myasthenic symptoms.
Beta-adrenergic receptor blocking agents may mask the signs and symptoms of acute hypoglycemia and clinical signs (eg, tachycardia) of hyperthyroidism. Use caution in patients subject to spontaneous hypoglycemia or in diabetic patients (especially those with labile diabetes) who are receiving insulin or oral hypoglycemic agents. Carefully manage patients who may develop thyrotoxicosis to avoid abrupt withdrawal of beta-adrenergic blocking agents that might precipitate a thyroid storm.
Ocular hypersensitivity has occurred with brimonidine tartrate ophthalmic solutions 0.2% (eg, increase in IOP).
Some authorities recommend gradual withdrawal of beta-adrenergic receptor blocking agents due to impairment of beta-adrenergically mediated reflexes during surgery. If necessary during surgery, the effects of beta-adrenergic blocking agents may be reversed by sufficient doses of adrenergic agonists.
ADVERSE REACTIONS
The most frequent reactions with COMBIGAN® in about 5% to 15% of patients included: allergic conjunctivitis, conjunctival folliculosis, conjunctival hyperemia, eye pruritus, ocular burning, and stinging.
DRUG INTERACTIONS
COMBIGAN® may reduce blood pressure. Use caution in patients on antihypertensives and/or cardiac glycosides.
Observe patients receiving a beta-adrenergic blocking agent either orally or intravenously and COMBIGAN® for additive effects of beta-blockade, both systemic and on intraocular pressure. Concomitant use of two topical beta-adrenergic blocking agents is not recommended.
Use caution in the co-administration of beta-adrenergic blocking agents (eg, COMBIGAN®) and oral or intravenous calcium antagonists due to possible atrioventricular conduction disturbances, left ventricular failure, and hypotension. Avoid co-administration in patients with impaired cardiac function.
Observe patients closely when a beta-blocker is administered to patients receiving catecholamine-depleting drugs (eg, reserpine) due to possible additive effects and the production of hypotension and/or marked bradycardia, which may result in vertigo, syncope, or postural hypotension.
Specific drug interaction studies have not been conducted with COMBIGAN®, but consider the possibility of an additive or potentiating effect with CNS depressants (alcohol, barbiturates, opiates, sedatives, or anesthetics).
Concomitant use of beta-adrenergic blocking agents with digitalis and calcium antagonists may have additive effects in prolonging atrioventricular conduction time.
Potentiated systemic beta-blockade (eg, decreased heart rate, depression) has been reported with combined use of CYP2D6 inhibitors (eg, quinidine, SSRIs) and timolol.
Tricyclic antidepressants (TCAs) can blunt the hypotensive effect of systemic clonidine. It is not known whether the concurrent use of TCAs with COMBIGAN® in humans can interfere with the IOP-lowering effect. Caution is advised in patients taking TCAs, which can affect the metabolism and uptake of circulating amines.
Monoamine oxidase (MAO) inhibitors may theoretically interfere with the metabolism of brimonidine and potentially increase systemic side effects such as hypotension. Use caution in patients taking MAO inhibitors, which can affect the metabolism and uptake of circulating amines.
Please see accompanying full Prescribing Information or visit https://www.rxabbvie.com/pdf/combigan_pi.pdf
DURYSTA® (bimatoprost intracameral implant) Important Information
INDICATIONS AND USAGE
DURYSTA® (bimatoprost intracameral implant) is indicated for the reduction of intraocular pressure (IOP) in patients with open angle glaucoma (OAG) or ocular hypertension (OHT).
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
DURYSTA® is contraindicated in patients with: active or suspected ocular or periocular infections; corneal endothelial cell dystrophy (e.g., Fuchs’ Dystrophy); prior corneal transplantation or endothelial cell transplants (e.g., Descemet’s Stripping Automated Endothelial Keratoplasty [DSAEK]); absent or ruptured posterior lens capsule, due to the risk of implant migration into the posterior segment; hypersensitivity to bimatoprost or to any other components of the product.
WARNINGS AND PRECAUTIONS
The presence of DURYSTA® implants has been associated with corneal adverse reactions and increased risk of corneal endothelial cell loss. Administration of DURYSTA® should be limited to a single implant per eye without retreatment. Caution should be used when prescribing DURYSTA® in patients with limited corneal endothelial cell reserve.
DURYSTA® should be used with caution in patients with narrow iridocorneal angles (Shaffer grade ˂ 3) or anatomical obstruction (e.g., scarring) that may prohibit settling in the inferior angle.
Macular edema, including cystoid macular edema, has been reported during treatment with ophthalmic bimatoprost, including DURYSTA® intracameral implant. DURYSTA® should be used with caution in aphakic patients, in pseudophakic patients with a torn posterior lens capsule, or in patients with known risk factors for macular edema.
Prostaglandin analogs, including DURYSTA®, have been reported to cause intraocular inflammation. DURYSTA® should be used with caution in patients with active intraocular inflammation (e.g., uveitis) because the inflammation may be exacerbated.
Ophthalmic bimatoprost, including DURYSTA® intracameral implant, has been reported to cause changes to pigmented tissues, such as increased pigmentation of the iris. Pigmentation of the iris is likely to be permanent. Patients who receive treatment should be informed of the possibility of increased pigmentation. While treatment with DURYSTA® can be continued in patients who develop noticeably increased iris pigmentation, these patients should be examined regularly.
Intraocular surgical procedures and injections have been associated with endophthalmitis. Proper aseptic technique must always be used with administering DURYSTA®, and patients should be monitored following the administration.
ADVERSE REACTIONS
In controlled studies, the most common ocular adverse reaction reported by 27% of patients was conjunctival hyperemia. Other common adverse reactions reported in 5%-10% of patients were foreign body sensation, eye pain, photophobia, conjunctival hemorrhage, dry eye, eye irritation, intraocular pressure increased, corneal endothelial cell loss, vision blurred, iritis, and headache.
Please see accompanying full Prescribing Information or visit https://www.rxabbvie.com/pdf/durysta_pi.pdf
LUMIGAN® (bimatoprost ophthalmic solution) 0.01% Important Information
INDICATION
LUMIGAN® 0.01% (bimatoprost ophthalmic solution) is indicated for the reduction of elevated intraocular pressure in patients with open angle glaucoma or ocular hypertension.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
LUMIGAN® 0.01% is contraindicated in patients with hypersensitivity to bimatoprost or to any of the ingredients.
WARNINGS AND PRECAUTIONS
Pigmentation
Bimatoprost ophthalmic solution has been reported to cause changes to pigmented tissues. The most frequently reported changes have been increased pigmentation of the iris, periorbital tissue (eyelid) and eyelashes. Pigmentation is expected to increase as long as bimatoprost is administered. After discontinuation of bimatoprost, pigmentation of the iris is likely to be permanent, while pigmentation of the periorbital tissue and eyelash changes have been reported to be reversible in some patients. Patients who receive treatment should be informed of the possibility of increased pigmentation. The long term effects of increased pigmentation are not known. Iris color change may not be noticeable for several months to years. While treatment with LUMIGAN® 0.01% can be continued in patients who develop noticeably increased iris pigmentation, these patients should be examined regularly.
Eyelash Changes
LUMIGAN® 0.01% may gradually change eyelashes and vellus hair in the treated eye. These changes include increased length, thickness, and number of lashes. Eyelash changes are usually reversible upon discontinuation of treatment.
lntraocular Inflammation
Prostaglandin analogs, including bimatoprost, have been reported to cause intraocular inflammation. In addition, because these products may exacerbate inflammation, caution should be used in patients with active intraocular inflammation (e.g., uveitis).
Macular Edema
Macular edema, including cystoid macular edema, has been reported during treatment with bimatoprost ophthalmic solution. LUMIGAN® 0.01% should be used with caution in aphakic patients, in pseudophakic patients with a torn posterior lens capsule, or in patients with known risk factors for macular edema.
Bacterial Keratitis
There have been reports of bacterial keratitis associated with the use of multiple-dose containers of topical ophthalmic products.
Contact Lens Use
LUMIGAN® 0.01% contains benzalkonium chloride, which may be absorbed by and cause discoloration of soft contact lenses. Contact lenses should be removed prior to instillation of LUMIGAN® 0.01% and may be reinserted 15 minutes following its administration.
ADVERSE REACTIONS
In a 12-month clinical study with bimatoprost ophthalmic solutions 0.01%, the most common adverse reaction was conjunctival hyperemia (31%). Approximately 1.6% of patients discontinued therapy due to conjunctival hyperemia. Other adverse drug reactions (reported in 1 to 4% of patients) with LUMIGAN® 0.01% in this study included conjunctival edema, conjunctival hemorrhage, eye irritation, eye pain, eye pruritus, erythema of eyelid, eyelids pruritus, growth of eyelashes, hypertrichosis, instillation site irritation, punctate keratitis, skin hyperpigmentation, vision blurred, and visual acuity reduced.
USE IN SPECIFIC POPULATIONS
Pediatric Use
Use in pediatric patients below the age of 16 years is not recommended because of potential safety concerns related to increased pigmentation following long-term chronic use.
Please see accompanying full Prescribing Information or visit https://www.rxabbvie.com/pdf/lumigan_pi.pdf
OZURDEX® (dexamethasone intravitreal implant) 0.7 mg Important Information
INDICATIONS AND USAGE
Diabetic Macular Edema
OZURDEX® (dexamethasone intravitreal implant) is a corticosteroid indicated for the treatment of diabetic macular edema.
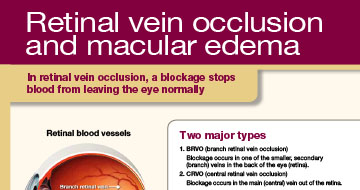
Retinal Vein Occlusion
OZURDEX® is a corticosteroid indicated for the treatment of macular edema following branch retinal vein occlusion (BRVO) or central retinal vein occlusion (CRVO).
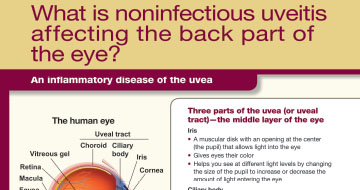
Posterior Segment Uveitis
OZURDEX® is indicated for the treatment of noninfectious uveitis affecting the posterior segment of the eye.
Dosage and Administration
FOR OPHTHALMIC INTRAVITREAL INJECTION. The intravitreal injection procedure should be carried out under controlled aseptic conditions. Following the intravitreal injection, patients should be monitored for elevation in intraocular pressure and for endophthalmitis. Patients should be instructed to report any symptoms suggestive of endophthalmitis without delay.
IMPORTANT SAFETY INFORMATION
Contraindications
Ocular or Periocular Infections: OZURDEX® (dexamethasone intravitreal implant) is contraindicated in patients with active or suspected ocular or periocular infections including most viral diseases of the cornea and conjunctiva, including active epithelial herpes simplex keratitis (dendritic keratitis), vaccinia, varicella, mycobacterial infections, and fungal diseases.
Glaucoma: OZURDEX® is contraindicated in patients with glaucoma, who have cup to disc ratios of greater than 0.8.
Torn or Ruptured Posterior Lens Capsule: OZURDEX® is contraindicated in patients whose posterior lens capsule is torn or ruptured because of the risk of migration into the anterior chamber. Laser posterior capsulotomy in pseudophakic patients is not a contraindication for OZURDEX® use.
Hypersensitivity: OZURDEX® is contraindicated in patients with known hypersensitivity to any components of this product.
WARNINGS AND PRECAUTIONS
Intravitreal Injection-related Effects: Intravitreal injections, including those with OZURDEX®, have been associated with endophthalmitis, eye inflammation, increased intraocular pressure, and retinal detachments. Patients should be monitored regularly following the injection.
Steroid-related Effects: Use of corticosteroids including OZURDEX® may produce posterior subcapsular cataracts, increased intraocular pressure, glaucoma, and may enhance the establishment of secondary ocular infections due to bacteria, fungi, or viruses.
Corticosteroids are not recommended to be used in patients with a history of ocular herpes simplex because of the potential for reactivation of the viral infection.
ADVERSE REACTIONS
Diabetic Macular Edema
Ocular adverse reactions reported by greater than or equal to 1% of patients in the two combined 3-year clinical trials following injection of OZURDEX® for diabetic macular edema include: cataract (68%), conjunctival hemorrhage (23%), visual acuity reduced (9%), conjunctivitis (6%), vitreous floaters (5%), conjunctival edema (5%), dry eye (5%), vitreous detachment (4%), vitreous opacities (3%), retinal aneurysm (3%), foreign body sensation (2%), corneal erosion (2%), keratitis (2%), anterior chamber inflammation (2%), retinal tear (2%), eyelid ptosis (2%). Non-ocular adverse reactions reported by greater than or equal to 5% of patients include: hypertension (13%) and bronchitis (5%).
Increased Intraocular Pressure: IOP elevation greater than or equal to 10 mm Hg from baseline at any visit was seen in 28% of OZURDEX® patients versus 4% of sham patients. 42% of the patients who received OZURDEX® were subsequently treated with IOP-lowering medications during the study versus 10% of sham patients.
The increase in mean IOP was seen with each treatment cycle, and the mean IOP generally returned to baseline between treatment cycles (at the end of the 6-month period).
Cataracts and Cataract Surgery: The incidence of cataract development in patients who had a phakic study eye was higher in the OZURDEX® group (68%) compared with Sham (21%). The median time of cataract being reported as an adverse event was approximately 15 months in the OZURDEX® group and 12 months in the Sham group. Among these patients, 61% of OZURDEX® subjects versus 8% of sham-controlled subjects underwent cataract surgery, generally between Month 18 and Month 39 (Median Month 21 for OZURDEX® group and 20 for Sham) of the studies.
Retinal Vein Occlusion and Posterior Segment Uveitis
Adverse reactions reported by greater than 2% of patients in the first 6 months following injection of OZURDEX® for retinal vein occlusion and posterior segment uveitis include: intraocular pressure increased (25%), conjunctival hemorrhage (22%), eye pain (8%), conjunctival hyperemia (7%), ocular hypertension (5%), cataract (5%), vitreous detachment (2%), and headache (4%).
Increased IOP with OZURDEX® peaked at approximately week 8. During the initial treatment period, 1% (3/421) of the patients who received OZURDEX® required surgical procedures for management of elevated IOP.
Please see accompanying full Prescribing Information or visit https://www.rxabbvie.com/pdf/ozurdex_pi.pdf
PRED FORTE® (prednisolone acetate ophthalmic suspension, USP) 1% sterile Important Information
INDICATIONS AND USAGE
PRED FORTE® (prednisolone acetate ophthalmic suspension, USP) 1% is indicated for the treatment of steroid-responsive inflammation of the palpebral and bulbar conjunctiva, cornea, and anterior segment of the globe.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
PRED FORTE suspension is contraindicated in acute untreated purulent ocular infections; in most viral diseases of the cornea and conjunctiva, including epithelial herpes simplex keratitis (dendritic keratitis), vaccinia, and varicella; and in mycobacterial infection of the eye and fungal diseases of ocular structures.
PRED FORTE suspension is also contraindicated in individuals with known or suspected hypersensitivity to any of the ingredients of this preparation and to other corticosteroids.
WARNINGS
Prolonged use of corticosteroids may result in posterior subcapsular cataract formation and may increase intraocular pressure in susceptible individuals, resulting in glaucoma with damage to the optic nerve and defects in visual acuity and fields of vision. Prolonged use may also suppress the host immune response and thus increase the hazard of secondary ocular infections.
If this product is used for 10 days or longer, intraocular pressure should be routinely monitored even though it may be difficult in children and uncooperative patients. Steroids should be used with caution in the presence of glaucoma. Intraocular pressure should be checked frequently.
Various ocular diseases and long-term use of topical corticosteroids have been known to cause corneal and scleral thinning. Use of topical corticosteroids in the presence of thin corneal or scleral tissue may lead to perforation.
Acute purulent infections of the eye may be masked or activity enhanced by the presence of corticosteroid medication.
The use of steroids after cataract surgery may delay healing and increase the incidence of bleb formation.
Use of ocular steroids may prolong the course and may exacerbate the severity of many viral infections of the eye (including herpes simplex). Employment of a corticosteroid medication in the treatment of patients with a history of herpes simplex requires great caution; frequent slit lamp microscopy is recommended.
PRED FORTE suspension contains sodium bisulfite, a sulfite that may cause allergic-type reactions, including anaphylactic symptoms and life-threatening or less severe asthmatic episodes in certain susceptible people. The overall prevalence of sulfite sensitivity in the general population is unknown and probably low. Sulfite sensitivity is seen more frequently in asthmatic than in non-asthmatic people.
PRECAUTIONS
General: The initial prescription and renewal of the medication order beyond 20 milliliters of PRED FORTE suspension should be made by a physician only after examination of the patient with the aid of magnification, such as slit lamp biomicroscopy, and, where appropriate, fluorescein staining. If signs and symptoms fail to improve after 2 days, the patient should be re-evaluated.
As fungal infections of the cornea are particularly prone to develop coincidentally with long-term local corticosteroid applications, fungal invasion should be suspected in any persistent corneal ulceration where a corticosteroid has been used or is in use. Fungal cultures should be taken when appropriate.
Information for Patients: Advise patients that if eye inflammation or pain persists longer than 48 hours or becomes aggravated, they should consult a physician.
Advise patients that to prevent eye injury or contamination, care should be taken to avoid touching the bottle tip to eyelids or to any other surface. The use of this bottle by more than one person may spread infection. Keep bottle tightly closed when not in use. Keep out of the reach of children.
Advise patients that PRED FORTE suspension contains benzalkonium chloride, which may be absorbed by soft contact lenses. Contact lenses should be removed prior to application of PRED FORTE and may be reinserted 15 minutes following its administration.
Pregnancy: Prednisolone has been shown to be teratogenic in mice when given in doses 1-10 times the human dose. Dexamethasone, hydrocortisone, and prednisolone were ocularly applied to both eyes of pregnant mice 5 times per day on days 10 through 13 of gestation. A significant increase in the incidence of cleft palate was observed in the fetuses of the treated mice. There are no adequate well-controlled studies in pregnant women. Prednisolone should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers: It is not known whether topical ophthalmic administration of corticosteroids could result in sufficient systemic absorption to produce detectable quantities in breast milk. Systemically administered corticosteroids appear in human milk and could suppress growth, interfere with endogenous corticosteroid production, or cause other untoward effects. Because of the potential for serious adverse reactions in nursing infants from prednisolone, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use: The safety and effectiveness in pediatric patients have been established. Use in pediatric patients is supported by evidence from adequate and well-controlled studies of prednisolone acetate ophthalmic suspension in adults with additional data in pediatric patients.
ADVERSE REACTIONS
The following adverse reactions have been identified during use of PRED FORTE. Because reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Adverse reactions include elevation of intraocular pressure (IOP) with possible development of glaucoma and infrequent optic nerve damage, posterior subcapsular cataract formation, and delayed wound healing.
The development of secondary ocular infection (bacterial, fungal, and viral) has occurred. Fungal and viral infections of the cornea are particularly prone to develop coincidentally with long-term applications of steroids. The possibility of fungal invasion should be considered in any persistent corneal ulceration where steroid treatment has been used.
Other adverse reactions reported with the use of prednisolone acetate ophthalmic suspension include allergic reactions; dysgeusia; eye pain; foreign body sensation; headache; pruritus; rash; transient burning and stinging upon instillation and other minor symptoms of ocular irritation; urticaria; and visual disturbance (blurry vision).
Keratitis, conjunctivitis, corneal ulcers, mydriasis, conjunctival hyperemia, loss of accommodation, and ptosis have occasionally been reported following local use of corticosteroids. Corticosteroid-containing preparations have also been reported to cause acute anterior uveitis and perforation of the globe.
Please see accompanying full Prescribing Information or visit https://www.rxabbvie.com/pdf/pred-forte_pi.pdf
RESTASIS® (cyclosporine ophthalmic emulsion) 0.05% and RESTASIS MULTIDOSE® (cyclosporine ophthalmic emulsion) 0.05% Important Information
INDICATIONS AND USAGE
RESTASIS® and RESTASIS MULTIDOSE® ophthalmic emulsion are indicated to increase tear production in patients whose tear production is presumed to be suppressed due to ocular inflammation associated with keratoconjunctivitis sicca. Increased tear production was not seen in patients currently taking topical anti-inflammatory drugs or using punctal plugs.
IMPORTANT SAFETY INFORMATION
Contraindications
RESTASIS® and RESTASIS MULTIDOSE® are contraindicated in patients with known or suspected hypersensitivity to any of the ingredients in the formulation.
Warnings and Precautions
Potential for Eye Injury and Contamination: Be careful not to touch the container tip to your eye or other surfaces to avoid potential for eye injury and contamination.
Use With Contact Lenses: RESTASIS® and RESTASIS MULTIDOSE® should not be administered while wearing contact lenses. If contact lenses are worn, they should be removed prior to the administration of the emulsion. Lenses may be reinserted 15 minutes following administration of RESTASIS® and RESTASIS MULTIDOSE® ophthalmic emulsion.
Adverse Reactions
In clinical trials, the most common adverse reaction following the use of cyclosporine ophthalmic emulsion 0.05% was ocular burning (upon instillation)—17%. Other reactions reported in 1% to 5% of patients included conjunctival hyperemia, discharge, epiphora, eye pain, foreign body sensation, pruritus, stinging, and visual disturbance (most often blurring).
Please see accompanying full Prescribing Information or visit https://www.rxabbvie.com/pdf/restasis_pi.pdf
Please see accompanying full Prescribing Information or visit https://www.rxabbvie.com/pdf/restasis-multidose_pi.pdf
VUITY® (pilocarpine hydrochloride ophthalmic solution) 1.25% Important Information
INDICATION
VUITY® (pilocarpine hydrochloride ophthalmic solution) 1.25% is indicated for the treatment of presbyopia in adults.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
VUITY is contraindicated in patients with known hypersensitivity to any ingredient in the formulation.
WARNINGS AND PRECAUTIONS
Miotics, including VUITY, may cause accommodative spasm. Patients should be advised not to drive or operate machinery if vision is not clear (e.g., blurred vision). In addition, patients may experience temporary dim or dark vision with miotics, including VUITY. Patients should be advised to exercise caution in night driving and other hazardous activities in poor illumination.
Rare cases of retinal detachment and retinal tear have been reported with miotics, including VUITY. Individuals with pre-existing retinal disease are at increased risk. Therefore, examination of the retina is advised in all patients prior to the initiation of therapy. Patients should be advised to seek immediate medical care with sudden onset of flashing lights, floaters, or vision loss.
VUITY is not recommended to be used when iritis is present because adhesions (synechiae) may form between the iris and lens.
Contact lens wearers should be advised to remove their lenses prior to the instillation of VUITY and to wait 10 minutes after dosing before reinserting their contact lenses.
To prevent eye injury or contamination, care should be taken to avoid touching the dispensing bottle to the eye or to any other surface.
ADVERSE REACTIONS
The most common adverse reactions (>5%) reported in clinical trials were headache, conjunctival hyperemia, and eye irritation.
Please see accompanying full Prescribing Information or visit https://www.rxabbvie.com/pdf/vuity_pi.pdf
XEN® Gel Stent (XEN® 45 Gel Stent preloaded into a XEN® Injector) Important Information
INDICATIONS
The XEN® Glaucoma Treatment System (XEN® 45 Gel Stent preloaded into a XEN® Injector) is indicated for the management of refractory glaucomas, including cases where previous surgical treatment has failed, cases of primary open-angle glaucoma, and pseudoexfoliative or pigmentary glaucoma with open angles that are unresponsive to maximum tolerated medical therapy.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
XEN® Gel Stent is contraindicated in angle-closure glaucoma where angle has not been surgically opened, previous glaucoma shunt/valve or conjunctival scarring/pathologies in the target quadrant, active inflammation, active iris neovascularization, anterior chamber intraocular lens, intraocular silicone oil, and vitreous in the anterior chamber.
WARNINGS
XEN® Gel Stent complications may include choroidal effusion, hyphema, hypotony, implant migration, implant exposure, wound leak, need for secondary surgical intervention, and intraocular surgery complications. Safety and effectiveness in neovascular, congenital, and infantile glaucoma has not been established. Avoid digital pressure following implantation of the XEN® Gel Stent to avoid the potential for implant damage.
PRECAUTIONS
Examine the XEN® Gel Stent and XEN® Injector in the operating room prior to use. Monitor intraocular pressure (IOP) postoperatively and if not adequately maintained, manage appropriately. Stop the procedure immediately if increased resistance is observed during implantation and use a new XEN® system. Safety and effectiveness of more than a single implanted XEN® Gel Stent has not been studied.
ADVERSE EVENTS
The most common postoperative adverse events included best-corrected visual acuity loss of ≥ 2 lines (≤ 30 days 15.4%; > 30 days 10.8%; 12 months 6.2%), hypotony IOP < 6 mm Hg at any time (24.6%; no clinically significant consequences were associated, no cases of persistent hypotony, and no surgical intervention was required), IOP increase ≥ 10 mm Hg from baseline (21.5%), and needling procedure (32.3%).
Caution: Federal law restricts this device to sale by or on the order of a licensed physician. Please click here for the full Directions for Use. Please call 1-800-433-8871 to report an adverse event.
Please see accompanying full Directions for Use or visit https://www.rxabbvie.com/pdf/xen_dfu.pdf
US-MULT-230264









